I’m a 4th year masters student studying at the University of Nottingham and my project is based on type 1 myotonic dystrophy (DM1) . I’m here to raise awareness of this condition and to highlight the importance of continued research in this field. I’ll begin by giving some brief background information. DM is a muscle wasting disease affecting around 1 in 8000 people. The picture below displays the key symptoms of DM. These include:
– muscle wasting
– cataracts
– myotonia (inability to relax voluntary muscles)
– cardiac conduction defects

In simple molecular terms, DM1 is caused by expansions of the CTG repeat in the dystrophia myotonica protein kinase (DMPK) gene; hence the name of the disease. The severity of the condition increases with the length of the expansions which can range from several hundred to several thousand, rather than the 5-35 seen in normal individuals. The most classical form is the adult-onset form. There is also the childhood-onset form and the most severe form which is congenital DM1.
Currently, there are no treatments available to slow the progression of DM. This is because the disease is very complex in nature and not fully understood. Therefore, there is significant need for further research to be conducted, which is why my project is so important. I hope that my results will highlight a greater understanding of the genes involved in this multicomplex disease.
The story so far
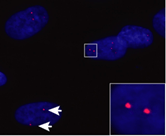
One key hallmark of the condition is the presence of distinct spots in the nucleus of DM1 cells and tissues. These spots are called nuclear foci and their accumulation is thought to cause the pathogenic nature of DM1. My project centres around these spots and uncovering genes that may be involved in their formation. To help visualise these spots I have included an example of the foci as found in previous research.
Genes of Interest
My genes of interest are components of the nonsense-mediated decay pathway. This is a surveillance pathway that acts to degrade mRNAs containing premature termination codons. The genes I’m focusing on are:
UPF1– core component of the nonsense-mediated decay pathway
SMG6– endonuclease that cleaves NMD targets resulting in the initiation of NMD-mediated degradation
The overall aim of the project is to knockout these two genes and determine if this has any effect on the formation of the nuclear foci in human DM1 cells.
CRISPR/Cas9
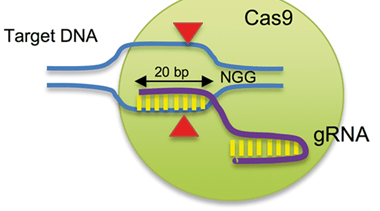
The gene editing tool I am using to knock out my genes of interest is called CRISPR/Cas9. This system consists of a single guide RNA (sgRNA) which binds to the complementary sequence of the genomic target (my gene of interest), and a Cas9 protein which makes a double stranded break in the DNA. This protein essentially acts as a pair of molecular scissors. The double stranded break it makes enables the knockout of my gene of interest. I have attached a Youtube video if you want to learn how this process works in more depth.
Challenges faced so far

Preliminary experiments showed me that the density the DM1 cells were plated at was an issue. I wanted to plate the cells at low density, however I found that the cells do not survive well at low density. I therefore explored the possibility of plating the transfected cells on feeder layers. Feeder layers are used to support cells by providing nutrient factors. I ultimately decided to plate the cells alone and with a layer of non-transfected cells acting as a feeder layer. So far I have not found any benefits to using the extra layer of non-transfected cells.
Fish and Chip event

I was lucky enough to attend the annual fish and chip event run by the Myotonic Dystrophy support group.
The founder, Margaret Bowler, was most amiable and chatted to us about the work she does. The charity aims to offer support and friendship to all those affected by myotonic dystrophy. As it is a fairly rare condition, families affected can often feel isolated . This charity allows them to come together and meet others affected by DM. It also has a leading role in raising awareness of the condition to the wider population.

At the event I was able to chat to a variety of people involved in the charity, including genetic counsellors. It is their job to give the families facts about the disease which will enable them to make decisions about their future, such as family planning and the routes to go down to reduce risk.
I also met Mike who was a trustee and treasurer of the charity. He told me about his wife and two sons who all suffer from the condition. It opened my eyes to the real life struggles that the disease can bring. Even little things like the inability to lift a kettle or open the lid of a coke bottle is taken away from you. Mike gave me a booklet about the challenges of DM sufferers undergoing anaesthetic operations which is something I hadn’t realised prior to the event. It was a humbling experience that further inspired me in the work that I am doing.

Contribution to future research
If by the end of my research I discover that my genes of interest do contribute to the formation of RNA foci in DM1 cells, this could help uncover new targets for future treatments. However, if this does not prove to be the case, there are plenty of other genes to explore in future research.
